Install the following C/C++ libraries: glpk, gsl and QuadProg++. If you cannot install those packages in the standard system directories but install them at
/your/installed/path/ for example, you have to modify the environment variablesLD_LIBRARY_PATH andCXXFLAGS by :export CXXFLAGS="-g -O3 -I/your/installed/path/include -L/your/installed/path/lib" export LD_LIBRARY_PATH="/your/installed/path/lib:"$LD_LIBRARY_PATH
If the compiler complains that it cannot find the library even you installed them by default, you should also find the installed path of these libraries and specify the two environment variables above manually. This may happen on QuadProg installed on Ubuntu by "apt-get install" command.Compile the
graphlib package in the source code. The compilation follows the standard configure, make process by executing the following commands in sequence:./configure make
-
Compile the
isoinfer package in the source code. The compilation follows the standard configure, make process by executing the following commands in sequence:./configure make
Note that it is necessary to keep the
Usage:
IsoInfer <Job> <Options>Jobs:
| |
Print help information |
| |
Extract junction ref sequence. In the involved parameters,
|
| |
Infer isoforms. In the involved parameters, |
Options:
| Parameter | Range | Default Value | Description |
| |
number | 0 | For job |
| |
file | N/A | Boundary file. The format of the file is :
chromosome strand position type
|
| |
file | N/A | Gene range file. The format of the file is :
gene_name chromosome strand start_position end_position
|
| |
file | N/A | TSS and PAS file. The format of the file is :
gene_name TSSs PASs
This parameter is optional. When this parameter is set, make sure that gene names are consistent with gene names
provided by parameter |
| |
file | N/A | Reference sequence in a single file. |
| |
file | N/A | A file storing the basic read information. The format of this file is:
mapping_file 0/1 [end_len] cross_strength noise_level total_read_cnt distribution_type definition_of_a_distributionThe format for the mapping_file is : chromosome strand start_positions end_positions
For example, a paired-end read with end length 50 is mapped to the RefSeq. The first end of this paired-end read is mapped to segments: [300,330) and [700,720) on the positive strand of the chromosome chr1 and the second end of this paired-end read is mapped to segments: [1300,1315) and [2000,2035) on the positive strand of the chromosome chr1. Then the mapping file for this paired-end read should contain two consecutive lines: chr1 + 300,700 330,720 chr1 + 1300,2000 1315,2035 The second term in a read info file indicates whether the read is
paired-end or not. If it is paired-end, set 1 here. Otherwise 0
should be set. If it is 1, then the first item in the following
line is the length of each end of the paired-end read. The lengths
of the two ends of a paired-end read is supposed to be the same. If
the read is not paired-end, then the following line starts
from In the second line of a read_info file,
After one read info, another one could be followed in the same file. On job -ext_junc_ref, only the first read info in the file is effective. If, on some job, not all the information in the read info is usefully, then the unused items can be set to any value. |
| |
T/F | F | Whether are the operations strand specific? |
| |
number | 0 | The minimum expression level in RPKM. By default, this parameter will not have influence on the result. The greater the value is, the higher sensitivity / lower precision the result achieves. For example, this parameter could be set as 1. |
| |
number | 3 | When doing job |
| |
number | 0 | Genes with expression levels (RPKM) below this parameter would be filtered out. A larger value for this parameter leads to a better precision. |
| |
number | 1 | A junction is covered if at least |
| |
number | 7 | Partition size. On whole mouse genome, the isoform inference process (Step4 in the following example) costs about 10 minutes on a standard PC with this default parameter. A larger value is supposed to lead to better results. |
| |
number in [0,1] | 0.05 | Set the confidence level. |
| |
file | N/A | A file for output
|
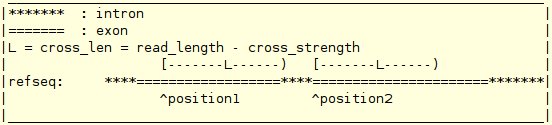
The following example is based on single-end short reads. In the following example, an example read_info file and several useful scripts are provided. The usages of all the scripts are straightforward. Please read the script for the usages.
- Generate files needed by
-bound ,-grange and-tsspas - Use a script knownGeneExtractor to
extract all the three files from a knownGene table downloaded from UCSC.
./knownGeneExtractor knownGene - Use a script BoundfromTopHat to
extract the file needed by
-bound from the predicted junctions by TopHat../BoundfromTopHat junctions.bed - Generate a read_info file
- If you have 10000000 single-end reads, read length is 50bp, read_mapping file is "your_mapped_reads", noise level is 0.1 RPKM and
cross_strength is 2. The content of the read_info file should be:
your_mapped_reads 0 2 0.1 10000000 0 50
- If you have 30000000 paired-end reads, end length is 30bp, read_mapping file is "your_mapped_reads", noise level is 0.1 RPKM,
cross_strength is 2 and the span (the gap plus 2 times of the end length) of the paired-end read follows normal distribution N(200,10^2). The content of the read_info file should be:
your_mapped_reads 1 30 2 0.1 30000000 1 200 10
- Generate a read mapping file
-
First, extract junction sequences non-strand-specifically using IsoInfer:
isoinfer -ext_junc_ref -s F -rstart 0 -bound Bound -grange GeneRange -tsspas TSSPAS -ref refseq -read_info read_info -o juncrefNote that the knownGene table (if it is used) and the reference sequence you downloaded should be consistent. Only the read length and cross strength in the read_info file is used in this step.Second, use Bowtie to map single-end short reads to the reference sequence and junction sequences. You can use the script tranMappedRefReads to extract the mapping information of reads to the reference sequence from the default output of bowtie. You can use the script tranMappedJuncReads to extract the mapping information of reads to junction sequences from the default output of bowtie. Then put the output of these two scripts together, e.g. into file "mapped_reads".
- Predict isoforms
Set
-min_exp to 0.1. All segments with expression level below 3 RPKM are considered as introns. A junction is covered if it is covered by at least 2 reads. Set all other parameters to default.isoinfer -predict -bound Bound -grange GeneRange -tsspas TSSPAS -intron_exp 3
-min_dup 2 -min_exp 0.1 -read_info read_info -o resultsThe TSS/PAS information is missing.
isoinfer -predict -bound Bound -grange GeneRange -read_info read_info -o results