IsoInfer is a C/C++ program to infer isoforms based on short RNA-Seq (single-end and paired-end) reads, exon-intron boundary and TSS/PAS information. This version of IsoInfer uses a unified way to handle different types of short reads with different lengths. The source code is provided for non-commercial usage. We appologize for the unavailability of the Windows and Mac versions of this program at the present time.
-
Install the following C/C++ libraries: glpk, gsl and QuadProg++. If you cannot install those packages in the standard system directories but install them at "/your/installed/path/" for example, you have to modify the environment variables LD_LIBRARY_PATH and CXXFLAGS by :
If the compiler complains that it cannot find the library even you installed them by default, you should also find the installed path of these libraries and specify the two environment variables above manually. This may happen on QuadProg installed on Ubuntu by "apt-get install" command.
$export CXXFLAGS="-I/your/installed/path/include -L/your/installed/path/lib"
$export LD_LIBRARY_PATH="/your/installed/path/lib:"$LD_LIBRARY_PATH
-
Compile the graphlib package in the source code. The compilation follows the standard configure, make process by execute the following commands in sequence:
$./configure
$make
-
Compile the isoinfer package in the source code. The compilation follows the standard configure, make process by execute the following commands in sequence:
$./configure
$make
Note that it is necessary to keep the graphlib and isoinfer under the same directory. Environment variable LD_LIBRARY_PATH should be exported like the first step every time when you open a new shell to run the program. I suggest you putting it in the .bashrc file in your home directory.
Usage:
IsoInfer <Job> <Options>Jobs:
| -h | Print help information |
| -ext_junc_ref | Extract junction ref sequence. -rstart, -bound, -grange, -tsspas, -ref, -read_info are required. |
| -gen_instance | Generate instances of problem for IsoInfer. Expression level will be used to define expressed segments. A segment is expressed if the expression level on this segment is above the noise level specified in the read_info file. -bound, -grange, -tsspas and -read_info are required. |
| -predict | Infer isoforms provided the instances generated by -predict. -ins, -conf_level, -minexp, -ps are required. |
Options:
| -rstart | number | For job -ext_junc_ref, the parameter specifies the start position of the first neocliotide of a chromosome. This parameter is to make sure that the coordinations used in the program is consistent with the coordinations provided by -bound, -grange and -tsspas. Default 0. |
| -bound | file | Boundary file. The format of the file is : chromosome strand position type
|
| -grange | file | Gene range file. The format of the file is : gene_name chromosome strand start_position end_position
|
| -tsspas | file | TSS and PAS file. The format of the file is : gene_name TSSs PASs
|
| -ref | file | Reference sequence in a single file. |
| -read_info | file | A file storing the basic read information. The format of this file is: mapping_file 0/1 [end_len] cross_strength noise_level total_read_cnt distribution_type "definition of a distribution" The format for the mapping_file is : chromosome strand start_positions end_positions
my_map_file 1 20 3 5 10000000 1 300, 30 After one read info, another one could be followed in the same file. On job -ext_junc_ref, only the first read info in the file is effective. If, on some job, not all the information in the read info is usefully, then the unused items can be set to any value. If there are more than one read infos in this file, the overall noise level is the maximum noise level among all the read infos. When doing job -gen_instance, a segment with expression level below the overall noise level will be considered as an intron. A carefully selected noise level is critical. If the noise level is 0, then all the segments will considered as expressed, which will introduce noise segments in the following isoform predictions. If the noise level is too high, many expressed segments will be considered as introns, which will lower the sensitivity. By our tests, 3~5 is a reasonable value for this parameter. |
| -update_read_info | T/F | Whether to update the read_info file by correcting the "total_read_cnt" in the read info file. This parameter is only effective when short reads are loaded. Default F |
| -s | T/F | Whether the operations are strand specific or not? Default F |
| -ins | file | A file containing instances. |
| -min_exp | number | The minimum expression level. Default 0. |
| -ps | number | Partition size. Default 7. On whole mouse genome, the isoform inference process (Step4 in the following example) costs about 10 minutes on a standard PC with this default parameter. A larger value is supposed to lead to better results. |
| -conf_level | number in [0,1] | Set the confidence level. Default 0.05. |
| -o | file | A file for output
|
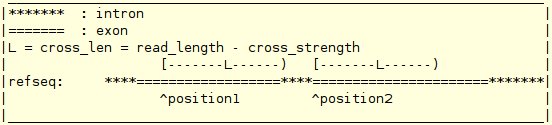
The following example is based on single-end short reads. In the following example, an example read_info file and several useful scripts are provided. The usages of all the scripts are straight forward. Please read the script for the usages.
- Use a script knownGeneExtractor to
extract the required files needed by -bound -grange and -tsspas from a
knownGene table downloaded from UCSC
$./knownGeneExtractor knownGene
- Modify the the example read_info file as you want.
- Extract junction sequences non-strand-specifically
$isoinfer -ext_junc_ref -s F -rstart 0 -bound Bound -grange GeneRange -tsspas TSSPAS -ref refseq -read_info read_info -o juncref
Note that the known gene table and the reference sequence you downloaded should be consistent. Only the read length and cross strength in the read_info file is used in this step. - Use Bowtie to map the short reads to the reference sequence and
junction sequences. You can use the script tranMappedRefReads to extract the
mapping information of reads to the reference sequence from the default output
of bowtie. You can use the script tranMappedJuncReads to extract the
mapping information of reads to junction sequences from the default output
of bowtie. Then put the output of these two scripts together, e.g. into file "mapped_reads".
- Modify the "mapping_file", "total_read_cnt" in the example read_info file. The "mapping_file" should
be "mapped_reads" and "total_read_cnt" should be the total number of mapped short reads including those mapped to the reference sequence
and to the junction sequences.
- Generate instances for IsoInfer and correct the total number of mapped reads in the read_info file automatically.
$isoinfer -gen_instance -bound Bound -grange GeneRange -tsspas TSSPAS -read_info read_info -update_read_info T -o my_instances - Predict isoforms given input 'my_instances'. Set -minexp to 1 and all other parameters to default.
$isoinfer -predict -ins my_instances -min_exp 1 -read_info read_info -o results
Jianxing Feng, Wei Li and Tao Jiang. Inference of isoforms from short sequence reads (Extended Abstract). 14th Annual International Conference on Research in Computational Molecular Biology (RECOMB), Lisbon, Portugal, April 25-28, 2010
Please email to:
jianxing
TA
cs.ucr.edu